
A Anvisa lançou a plataforma Business Intelligence (BI) da fila completa de produtos de diagnóstico in vitro para Covid-19. A plataforma de inteligência empresarial proporciona transparência à situação dos pedidos protocolados na Agência.
A novidade promove a consulta a diversos dados, como por exemplo a quantidade de pedidos deferidos, indeferidos, em análise, aguardando o certificado de Boas Práticas de Fabricação (CBPF), entre outros. Acesse o BI de produtos de diagnóstico in vitro para Covid-19 ou acompanhe a seguir:
Destaca-se que, desde o dia 18 de março, a Agência já avaliou mais de 120 pedidos de registro de produtos para testagens relacionadas à Covid-19. A maior parte das petições aguarda complementação de informações por parte das empresas e outras estão sendo analisadas com prioridade.
O tempo médio para avaliação dos registros pela Anvisa está em torno de 15 dias. Confira a relação dos produtos para diagnóstico in vitro de Covid-19 regularizados.
Prioridade No dia 18 de março deste ano, a Anvisa publicou a Resolução da Diretoria Colegiada (RDC) 348/2020, que estabeleceu regras extraordinárias e temporárias para agilizar a avaliação de novos produtos, por meio da priorização da análise de pedidos de registro de testes para detecção do novo coronavírus.
A medida faz parte das ações estratégicas para viabilizar produtos que possam ser utilizados no enfrentamento da pandemia de Covid-19 de forma mais ágil e com a manutenção dos critérios de segurança e eficácia.
Atuação pré e pós-mercado
O controle do risco sanitário envolve ações pré e pós-mercado, ou seja, que acontecem antes do produto ser comercializado e que decorrem do uso e monitoramento do comportamento do produto após a sua comercialização.
O registro é parte da atuação do controle sanitário que ocorre antes que o produto seja comercializado no mercado nacional. A partir dele pode-se verificar se as empresas envolvidas nos processos fabris e nas atividades de importação encontram-se habilitadas para a realização destas atividades.
Os produtos para diagnóstico in vitro da Covid-19 são de uso profissional e classificados como de alto risco aos indivíduos e à saúde pública (classe III), e as empresas que os fabricam devem atender aos critérios de Boas Práticas de Fabricação estabelecidos na RDC 16/2013. A Anvisa certifica as unidades fabris que cumprem os requisitos indicados nessa resolução que estão relacionados ao sistema de qualidade das empresas e aos controles aplicados no desenvolvimento e na fabricação dos produtos.
Além disso, o registro também engloba a avaliação de desempenho dos produtos. Para tanto, são apresentadas informações, na forma de um dossiê técnico documental, que permitem a avalição da confiabilidade dos resultados e da efetividade diagnóstica do produto. Os fabricantes precisam demonstrar como foram realizados os testes de desempenho e a qualificação das amostras utilizadas, além de evidências clínicas, tendo em vista o tipo de produto, a indicação de uso e a metodologia. Alguns estudos que fazem parte do dossiê técnico descrito na RDC 36/2015 estão indicados na figura abaixo:
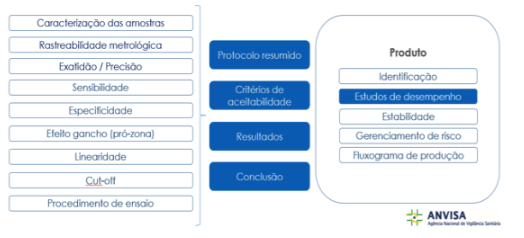
Durante o processo de registro de cada produto são verificadas as rotulagens e as instruções de uso, para que contemplem as informações essenciais para a sua utilização segura e orientem o profissional de saúde quanto aos procedimentos para execução do ensaio, o tipo de amostra, a interpretação dos resultados, as limitações e as advertências, entre outras informações relevantes.
No Brasil, o registro de produto importado deve ser feito por uma empresa nacional que tenha autorização do fabricante para representá-lo no país. Essa documentação também é parte do processo de avaliação.
A concessão do registro, portanto, é a primeira etapa do controle sanitário. É responsabilidade dos fabricantes e importadores disponibilizarem no mercado produtos que estejam em estrita conformidade com as informações aprovadas no registro. A Anvisa e as Vigilâncias Sanitárias estaduais e municipais continuam monitorando o comportamento dos produtos após a sua comercialização, seja por meio de queixas técnicas ou pela avaliação laboratorial de desempenho.
Estudos científicos
A comunidade científica vem estudando intensamente o comportamento do novo coronavírus (Sars-CoV-2) e atualmente apenas os ensaios que detectam antígenos são indicados para determinação do diagnóstico, pois permitem verificar se o vírus está presente na amostra testada. A Organização Mundial da Saúde (OMS) indica o ensaio molecular de RT-PCR como a referência (padrão ouro) para confirmação de casos de Covid-19.
Apesar de ainda existir muita incerteza em relação a esse novo vírus, a utilização de testes de anticorpos, aliada ao acompanhamento clínico, pode, ao longo do tempo, fornecer à comunidade médica mais informações sobre a recuperação de pacientes e sobre o risco de infeção pela exposição ao vírus, além de auxiliar na definição de políticas públicas.
Sendo assim, a Anvisa esclarece que pode haver situações em que a distribuição dos processos para análise pela Agência não atenda exclusivamente ao critério cronológico, sendo também consideradas as informações preferenciais quanto à metodologia RT-PCR e à unidade fabril/origem dos produtos. Esses três fatores combinados permitem otimizar a capacidade de avaliação pelo corpo técnico da instituição e ampliar a oferta de produtos para o diagnóstico da Covid-19.
Fonte: ANVISA
Foto: Cecília Bastos/USP IMAGENS&nbs





